




La CPAM è una rara malformazione del tratto respiratorio inferiore. Si tratta di una massa cistica o solida di tessuto polmonare, che è il risultato di un arresto di maturazione dei bronchioli terminali che proliferano in maniera anomala e della soppressione dello sviluppo degli alveoli. Generalmente interessa un lobo polmonare.
La CPAM ha un’incidenza di circa 1:25000-1:35000 casi, e rappresenta la più frequente patologia polmonare riscontrata all’esame ecografico di routine a 20 settimane di gravidanza.
La CPAM viene agevolmente diagnosticata mediante l’esame ecografico in cui appare come un’area iperecogena disomogenea con all’interno possibili aree cistiche anecogene di diverse dimensioni, contenenti liquido. Tra le diverse classificazioni esistenti, quella attualmente più utile in ambito ecografico soprattutto per il suo significato prognostico, distingue le forme di CPAM in macrocistiche e microcistiche.
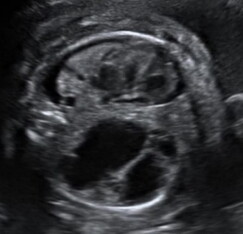
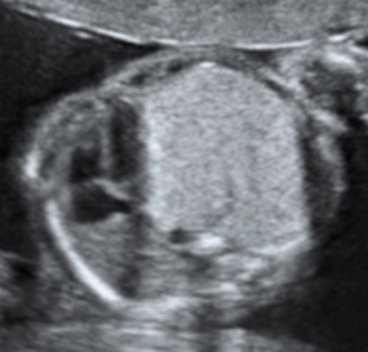
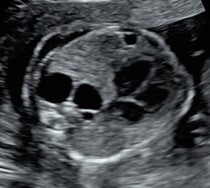
Immagini ecografiche di CPAM di tipo macrocistico (A), microcistico (B) e misto (C)
La CPAM è frequentemente un reperto isolato, oppure più raramente associarsi ad anomalie cardiache o cromosomiche. Se la CPAM è molto grande può determinare compressione dei vasi toracici causando nei casi più severi scompenso cardiaco e idrope, ossia accumulo di liquido all'interno del feto (ad esempio nel torace o nell’addome). La Risonanza Magnetica rappresenta un ottimo strumento di aiuto nello studio della morfologia e del volume polmonare.
La storia naturale della CPAM è caratterizzata nella maggior parte dei casi da un aumento della massa fino a 26-28 settimane. L’aumento eccessivo delle dimensioni della massa o la comparsa di complicanze quali il polidramnios e l’idrope fetale possono mettere a rischio il feto. In particolare, l’idrope rappresenta il segno prognostico più severo e rappresenta la principale indicazione all’intervento in utero.
Le opzioni terapeutiche per il trattamento della CPAM in utero prevedono diversi tipi di approccio a seconda che la CPAM sia micro o macro-cistica. Nel caso di CPAM microcistica o mista la terapia di elezione è quella medica, mediante l’utilizzo di corticosteroidi somministrati alla mamma.
Nei casi di grosse cisti che spostano il cuore e i vasi toracici, determinando la comparsa di idrope fetale, l’intervento ad oggi considerato più efficace consiste nel posizionamento per via eco-guidata, di un piccolo tubo di drenaggio tra la cavità cistica e quella amniotica (shunt toraco-amniotico), con lo scopo di decomprimere la cisti e migliorare lo stato emodinamico del feto.
Lo shunt toraco-amniotico è un intervento mininvasivo eseguito in anestesia locale materna, con un ricovero di circa 2 giorni, gravato da un rischio di rottura prematura delle membrane di circa il 10%.
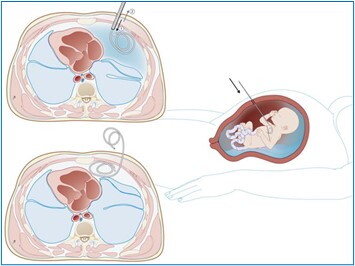
Figura: Posizionamento dello shunt toraco-amniotico che serve a decomprimere grosse cisti polmonari che possono comprimere il cuore e i vasi toracici fetali.
Nella maggior parte dei casi alla nascita il neonato è asintomatico e viene trasportato al Nido. Nei casi invece complicati, nel caso di difficoltà respiratoria, viene portato in Terapia Intensiva Neonatale dove viene assistito in maniera più adeguata, in quanto questi pazienti hanno bisogno di assistenza intensiva complessa e avanzata.
Nel caso di bambino asintomatico, anche quando la malformazione non era più visibile nell’ultimo periodo della gravidanza, verrà programmata una TAC del torace con mezzo di contrasto intorno ai 3 mesi di vita per poi programmazione dell’intervento chirurgico per rimozione della massa residua.
Iscriviti alla newsletter per ricevere i consigli degli specialisti del Bambino Gesù
- A cura di: Isabella Fabietti
Unità Operativa di Medicina e Chirurgia Fetale - in collaborazione con: